
pymol est un outil de visualisation graphique dédié aux structures moléculaires. Les jupyter notebooks sont des outils formidables pour organiser une succession d’étapes de calculs. Pouvoir utiliser ces deux outils ensemble peut être très avantageux.
Une instance de pymol par notebook
En définissant la classe suivante, il devient possible de lancer une instance de pymol pour chacun des notebooks utilisés. Attention , dès que le kernel est arrêté, la session de pymol associée disparaît.
class pymolPy3:
def __init__(self, mode = 1):
# Enable gui...
cmd = 'pymol -p'
# Turn off gui...
if mode != 1: cmd = 'pymol -pc'
# Create a subprocess...
self.pymolpy3 = Popen(cmd, shell=True, stdin=PIPE, stdout=DEVNULL)
def __del__(self):
# Turn off stdin...
self.pymolpy3.stdin.close()
# Wait until program is over...
self.pymolpy3.wait()
# Terminate the subprcess...
self.pymolpy3.terminate()
def do(self, s):
# Keep reading the new pymol command as a byte string...
res = self.pymolpy3.stdin.write( bytes( (s + '\n').encode() ) )
# Flush it asap...
res = self.pymolpy3.stdin.flush()
def get_Mp(
self
):
logger.debug("get_p_from_pymol")
# --------------------------------------------------------------------------
self.do(f"""
import sys
import numpy as np
sys.path.insert(0, f"/home/krezel@ICOA-03.LOCAL/Documents/Programmes/Gitlab")
from tools_SBC.basic import (get_path, ya, yapa, load_var, dump_var, bash)
Mp = cmd.get_coords("(sele)",1)
dump_var(Mp, "Mp")
""")
sleep(0.5)
return load_var("Mp")
En mettant ce code dans un fichier pymol.py, il devient possible de faire la chose suivante. On suppose que le répertoire courant possède un fichier protein.pdb d’une protéine.
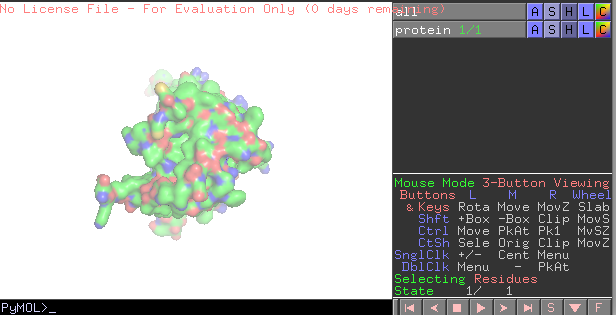
from pymol import pymolPy3
pm = pymolPy3()
path = get_path()
pm.do(f"""
cd {path}
delete all
bg_color white
load protein.pdb
util.cbag protein
show surface, protein
hide cartoon
set transparency, 0.5, protein
zoom
""")Les fonctions de ce type sont dans pymol.py
Une instance de pymol commune à tous les notebooks
Dans le cas où l’on veut travailler avec une seule instance de pymol qui soit commune à tous les notebooks, il faut lancer pymol à partir d’une console à l’aide de pymol -R
Ensuite il est possible de développer des fonctions comme la suivante qui permet d’afficher des molécules rdkit.
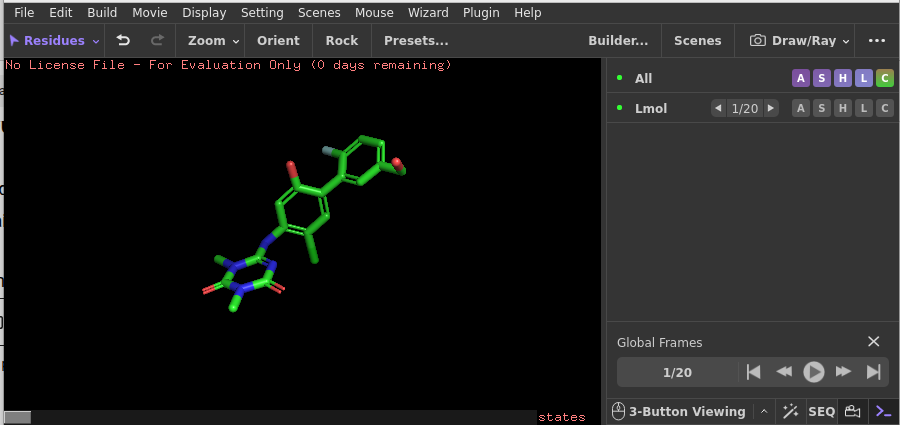
def pymol_show_Lmol(
Lmol,
name=None,
color="green",
style="stick",
cartoon=-1,
surface=False,
ref=None,
):
"""
"""
logger.debug("pymol_show_Lmol")
# --------------------------------------------------------------------------
cmdx = get_cmd()
path = get_path()
cmdx.do(f"cd {path}")
cmdx.do("set all_states, off")
if name is None:
name = get_s_from_object(Lmol)
fname = f"{name}.sdf"
create_sdf_from_Lmol(Lmol, fname, keep_prop=False)
pymol_load(
fname=fname,
name=name,
style=style,
color=color,
cartoon=cartoon,
surface=surface,
ref=ref,
)Les fonctions de ce type sont dans pymol_SCARTEK.py
Accéder à pymol depuis un jupyter lab lancé dans un docker
Nous sommes dans ls situation où jupyter lab fonctionne dans un docker. Pour que pymol puisse être vu , il faut taper la commande suivante:
export PYMOL_RPCHOST=0.0.0.0puis lancer pymol en mode RPC:
pymol-oss.pymol -R