
Accélération des calculs avec Networkx
Le module Networkx est un outil particulièrement intéressant pour traiter des problèmes associés aux graphes. Or les outils développés avec ce module deviennent souvent lents quand les graphes deviennent trop grands. Cependant ce problème est en train d’être résolu par NVIDIA comme l’explique cette vidéo.
Analyse des résultats expérimentaux dans la Chembl
Introduction
La base de donnée nommée Chembl est vraiment très intéressante pour essayer de comprendre les mécanismes en jeu lors de l’interaction entre un ligand et une protéine. En effet, sur le site de la Chembl, on trouve un lien vers les résultats expérimentaux liés aux cibles biologiques. En bas à gauche sur l’image ci-dessous.
En triant les cibles par nombre de molécules (compounds) testées sur une cible, on constate que certaines cibles ont été vraiment très étudiées:
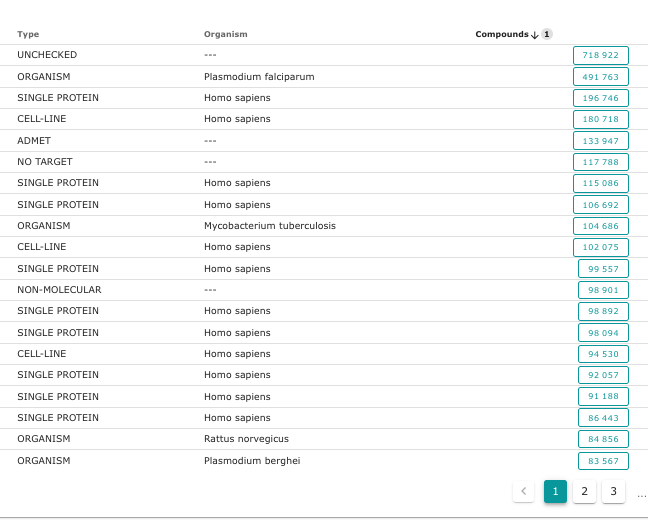
Ce qui nous intéresse en premier lieu, ce sont les résultats expérimentaux sur des “single proteins”, des protéines prises seules.
Pour commence une première analyse, j’ai choisi une protéine avec quelques milliers de molécules testées sur celle-ci, la CHEMBL3880, la “Heat shock protein HSP 90-alpha” avec 3019 molécules dont l’identifiant UniProt est P07900.
Un filtre sur le nombre maximum d’atomes (naat_max=30) a été appliquée et seules les molécules avec uniquement des atomes de chimie organiques ont été conservées soit 1855 molécules.
Le docking de ces molécules sur le site d’un inhibiteur dans le pdb 2YES donne le résultat suivant:
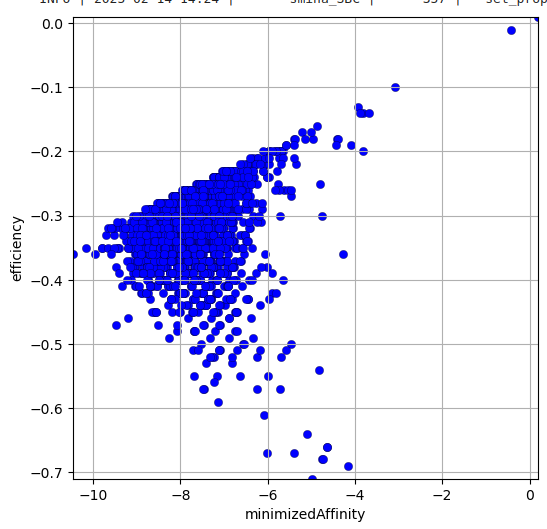
Molécules ayant une valeur de “‘minimizedAffinity” inférieure à -9,75:
 IC50 = 1700 nM IC50 = 1700 nM | 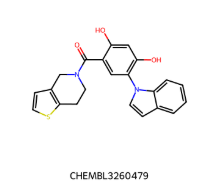 IC50 = 330 nM IC50 = 330 nM |
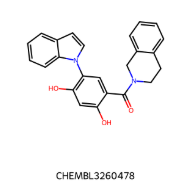 IC50=130 nM IC50=130 nM | 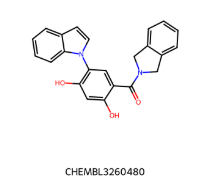 IC50 = 38 nM IC50 = 38 nM |
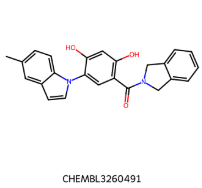 IC50=38 nM IC50=38 nM |
La molécules ayant la meilleure activité est la suivante:
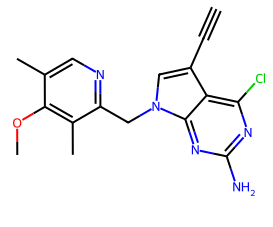 CHEMBL2205245 avec un IC50 de 1nM mais minimizedAffinity: -7.5 et efficiency: -0.31 CHEMBL2205245 avec un IC50 de 1nM mais minimizedAffinity: -7.5 et efficiency: -0.31 |
Différence de conformation entre CHEMBL3360305 et CHEMBL2205245
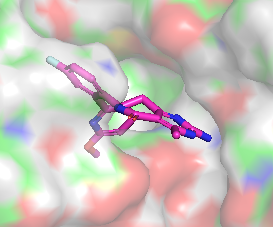 | 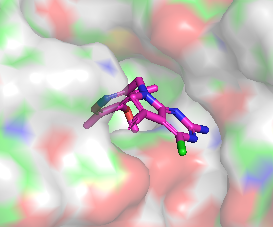 |
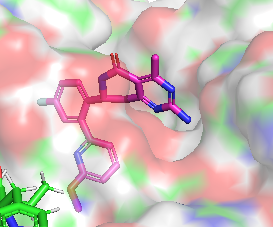 | 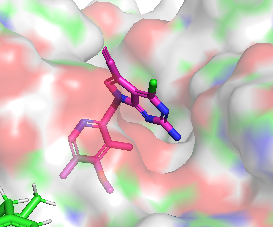 |
Recherche de cas simples avec beaucoup de données pour tester les DNN
Question: Est-il possible de trouver des exemples d’applications des DNN où les données sont abondantes ou peuvent être générées rapidement ?
La spectrométrie de masse des peptides (enchaînement de moins de 30 acides aminés) pourrait être une possibilité. Dans la base de données MassIVE, on trouve plus de 40 M de spectres de masses de peptides!
Simplification de la recherche des pKa d’une molécule donnée.
Par Pascal KREZEL, ICOA, le 12/02/2025
Les modèles actuels de prédiction de pKa cherchent à définir un pKa pour tous les sites d’une molécule. Or expérimentalement, une molécule possède moins de pKa que de fonctions acido-basiques. L’exemple suivant illustre ce problème. Dans SAMPL6 pKa Challenge, on trouve par exemple la molécule suivante avec 2 pKa.
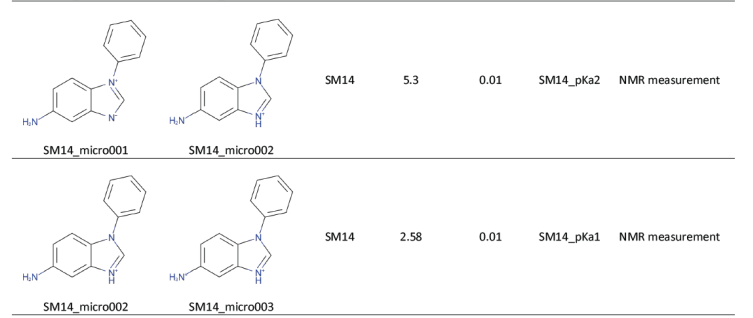
Si on demande à molGpKa, une prédiction des pKa, sa réponse est la suivante:
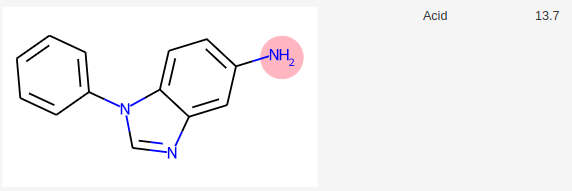
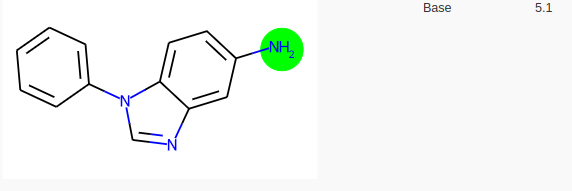
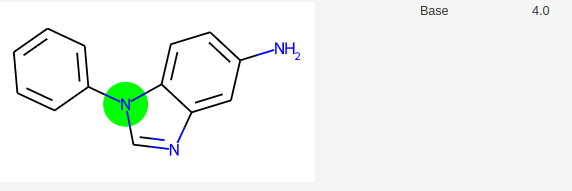
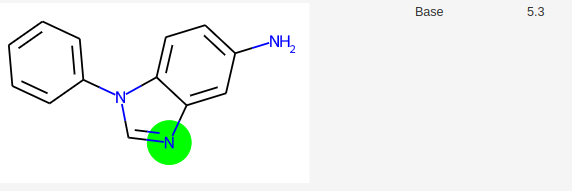
MolGpKa prédit donc 4 pKa.
Pour essayer de résoudre ce problème, nous proposons une nouvelle méthode passant par la prédiction d’une seule valeur, la charge moyenne à un pH donné.
Soit le cas extrême d’une molécule possédant 4 pKa dont il faut prédire les valeurs. Supposons qu’elle ait 2 fonctions acides et 2 fonctions basiques soit les équilibres suivants:
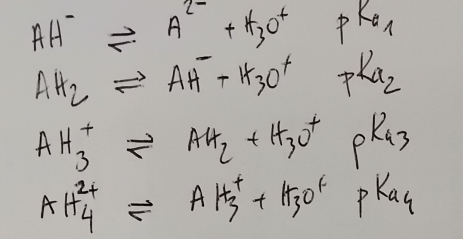
A pH fixé, on a les équations suivantes:
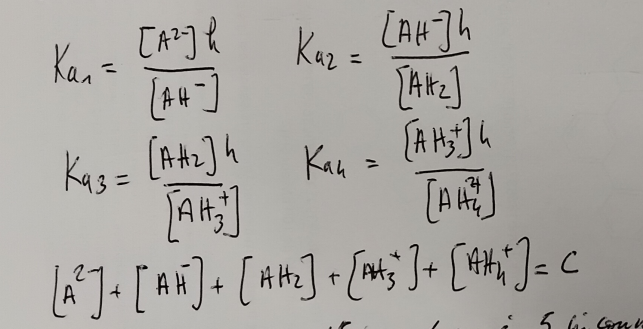
Ceci nous permet d’en déduire la charge moyenne à un pH donné:
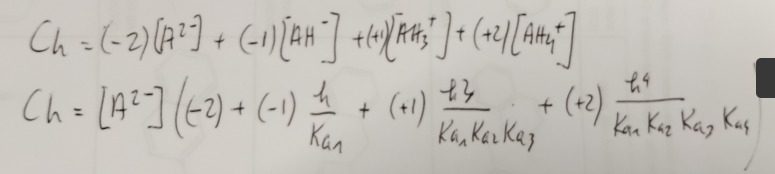
On en déduit une valeur de Ch dépendant uniquement de h et de C, la concentration totale.
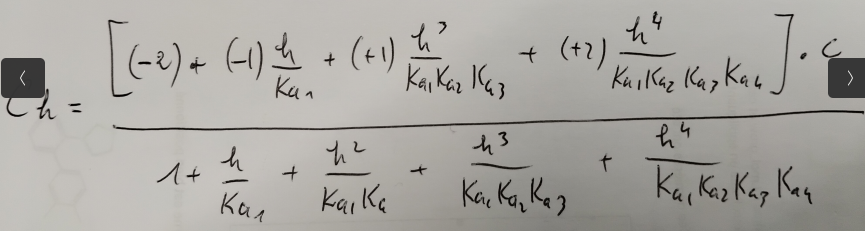
Finalement en fixant C à 1M, avec 4 valeurs de Ch pour différentes valeurs de pH, il est possible d’en déduire la valeur des différents Ka.
Si le système ne possède pas de solutions approximatives ou si les valeurs de Ka ne sont pas sont aberrantes, il faut en déduire que l’hypothèse des 2 fonctions acides et des 2 fonctions basiques était fausse et en prendre une autre.
Ainsi, en faisant au moins 4 modèles de prédiction de Ch pour différentes valeurs de pH, on doit être en mesure d’en déduire les différents pKa d’une molécule ayant au plus 4 pKa.
2025/01/23 : Rencontre avec les numériciens de l’ICMN
Suite à la demande de Jean-François Bardeau (JFB), Directeur de l’ICMN, nous (YS, PK et EE) avons rencontré les personnes de l’ICMS travaillant sur des problèmes numériques. JFB a commencé la réunion en souhaitant faciliter des échanges sur des problèmes de programmation potentielles. Nous avons présenté notre vision du projet SPGoO visant la valorisation des codes développés au sein des laboratoires de recherche et des données associées ainsi que le partage de bonnes pratiques pour rendre le travail de chacun plus efficace. 5 numériciens de l’ICMN étaient présents, ces derniers ont indiqué leur soucis de rendre leurs codes plus visibles sur Internet. Notre démarche leur semblait intéressante. Nous attendons leur retour suite à la lecture du site SPGoO pour commencer un éventuel premier projet.
2025/01/28: Rencontre pour labellisation d’un projet Cosmetic Valley
Yvan Stroppa a participé à la présentation du projet de recherche proposé par Patrick Baril (CBM) dans le cadre des appels à projets “Cosmetic Valley”. Ce projet comporte une part importante d’analyse de données approfondis (machine learning) que Patrick Baril souhaite réalisé avec l’aide d’Yvan Stroppa. La présence de dernier a permis de consolider le projet auprès du jury.
2024/11/22 : Présentation de SPGoO dans le cadre du CDL
Présentation de la cellule mutualisée : lien
2023 – Participation “4. International JSXgraph conference”
Présentation du projet JED : lien
Présentation de Swarm et K8S (06/2024)
Dans le cadre des mardis de l’informatique, présentation de solution d’orchestration de type Swarm et Kubernetes.
Intervention : Y. Stroppa
27 Juin 2024 (lien)